学术动态
R969.1
静脉注射给药的甘草酸易在由肝转运体OATP1B1/OATP1B3介导的药物相互作用中成为被影响药物
原文作者:董佳佳1,2,Olajide E. Olaleye1,姜蓉嵘1,李静1,吕闯3,杜飞飞1, 徐方1,杨军令1,王凤清1,贾伟伟1,李川1,2
1中国科学院上海药物研究所新药研究国家重点实验室,上海 201203,中国;2中国科学院大学,北京 100049,中国;3Department of DMPK,Sanofi,Cambridge,MA 02451,美国
董佳佳,女,博士研究生,研究方向:中药药代动力学。
Tel:021-20231000转1505; E-mail:sjgg1031@163.com
贾伟伟,通信作者,男,副研究员,研究方向:中药药代动力学。
Tel:021-20231000转1507; E-mail:chengchen@simm.ac.cn
李川,通信作者,研究员,博士生导师,研究方向:中药药代动力学。
Tel:021-50803106; E-mail:chli@simm.ac.cn
随意使用天然产品可能严重干扰患者正在接受的化药治疗,此已引起国际广泛关注。在中国中西医结合治疗疾病很普遍,研究中药与化药合用时发生药代性质药物相互作用风险,对于保证临床用药的有效性和安全性十分重要。不同于美英等西方国家,中国将中药作为药物来监管和使用,因此研究中药与化药合用的风险既涉及“中药影响化药”,也涉及“化药影响中药”。国际上围绕化药影响天然产品的研究还很少,这是因为与健康相关的天然产品在西方国家通常不具备药物身份,缺乏相应的药代动力学研究和技术。研究“化药影响中药”的风险首先应明确成分体内暴露改变能够影响中药的有效性或安全性,其重点是要搞清楚与他药合用能否通过某种机制改变这些成分的体内暴露。甘草酸是豆科Glycyrrhiza属中药甘草的主要活性成分,甘草酸二铵注射液具有保肝和抗炎作用,长期用药能降低慢性病毒性肝炎向肝硬化和肝癌转化的概率,在中国的肝病治疗中广泛应用。然而,高剂量长期使用甘草酸二铵注射液可引发假醛固酮增多症,出现高血压、低血钾症及外周水肿等毒副作用,其作用机理涉及甘草酸通过抑制11β-类固醇脱氢酶-2阻止肾中氢化可的松转化为可的松。研究表明:假醛固酮增多症的发生与甘草酸的血浆药时曲线下面积(AUC)增大密切相关。
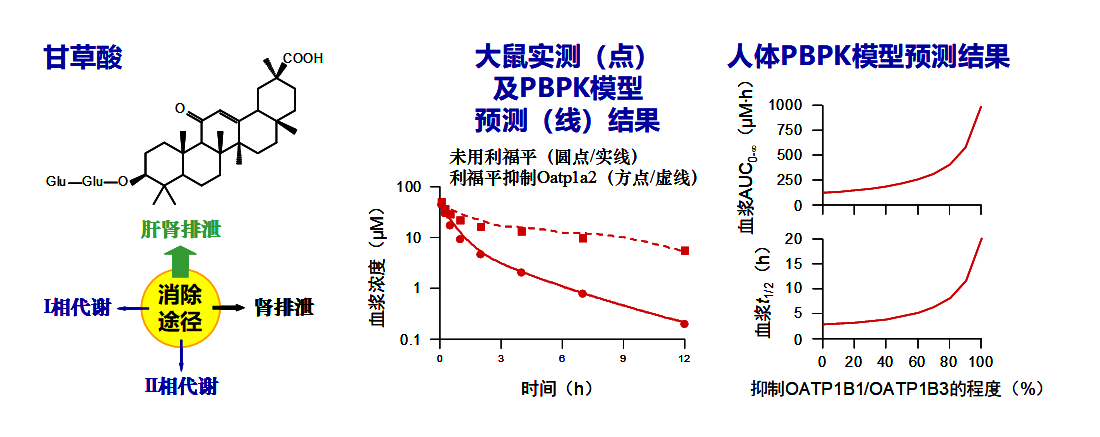
在本项研究中,采用转染细胞和膜囊泡技术,考察目前已知在人及大鼠肝细胞上所有的药物转运体中哪些能参与甘草酸的肝胆排泄,获取相关动力学数据。利用利福平抑制大鼠的肝转运体Oatp1b2,考察该转运体对静注给药后甘草酸系统暴露水平的影响。通过文献查阅和体内外实验研究获取甘草酸的药代动力学及相关数据,再利用生理药代动力学(PBPK)技术对其进行模拟并构建模型,应用该模型预测抑制人体肝转运体OATP1B1/OATP1B3对静注给药后甘草酸系统暴露水平的影响。
研究结果表明:静注给药后甘草酸主要以原型形式在血中暴露,甘草酸主要通过肝胆排泄从体循环中消除,通过代谢和肾排泄途径消除很慢。由于膜通透性差,甘草酸的肝胆排泄需借助肝转运体的作用,其作用机制是:血中甘草酸通过转运体OATP1B1/OATP1B3(人)或Oatp1b2(大鼠)的作用摄入肝细胞,随后再通过转运体MRP2/ABCP/BSEP/MDR1(人)或Mrp2/Abcp/Bsep(大鼠)的作用外排至胆汁。此外,转运体MRP3/MRP4(人)或Mrp4(大鼠)可将部分甘草酸从肝细胞外排至血中,通过与上述摄取转运体的互动,使肝胆排泄更加有效。在大鼠体内,一旦Oatp1b2的转运作用被抑制掉,甘草酸的血浆AUC就会显著增大、半衰期t1/2显著延长。PBPK模型预测显示:抑制OATP1B1/OATP1B3可使静注给药后甘草酸在人体内的血浆AUC增大、t1/2延长;当抑制程度超过80%时甘草酸的暴露改变尤为明显,且在连续给药时可产生蓄积,由此进一步增大AUC。
临床上有许多药物都是OATP1B1/OATP1B3的双重抑制剂,本项研究表明:这些药物与静注给药的甘草酸一同使用可引发药物相互作用,进而增大患者出现甘草酸假醛固酮增多症的风险。日本学者的临床研究提示:当与抗病毒药物帕利瑞韦、利托那韦及奥比他韦一同使用时,人体中甘草酸的血浆AUC0-24h可增大49%。有研究表明:帕利瑞韦和利托那韦均是OATP1B1/OATP1B3的双重抑制剂。需要指出的是:由于不同给药途径下体内暴露形式的不同,不能将静注给药的甘草酸制剂的药物相互作用风险及作用机理简单套用于口服给药。
找准中药与其他药物发生药代性质药物相互作用风险,对于中药临床合理用药十分重要。为此,需要从临床用药出发正确判定中药在相互作用中扮演的角色(“影响其他药物”或“被其他药物影响”),明确与相互作用关联的中药物质体内暴露特征及体内过程的关键环节,并明确相互作用的靶标和机理。由于在临床上大量使用,甘草酸制剂与其他药物发生相互作用的风险一直被人们所重视。之前开展的研究都是关注甘草酸“影响其他药物”的风险,涉及的作用机制包括:通过抑制OATP1B1/1B3及口服给药后通过诱导细胞色素P450酶的CYP3A4亚酶等。由于血浆蛋白结合率高,因此给药后甘草酸在体内没有足够的游离浓度对OATP1B1/1B3产生显著的抑制作用[相对靶向释药指数(DDI)=0.008-0.03]。由于口服后甘草酸很难在肠道吸收,需经代谢转化后进入体循环,因此口服给药后甘草酸诱导CYP3A4的物质基础和作用尚待进一步考察。本项研究系统揭示了静注给药后甘草酸的体内暴露特征及肝胆排泄的分子作用机理,发现了第一个具有临床意义且物质基础和作用机理清楚的甘草酸静注制剂与其他药物发生相互作用的风险,在相互作用中甘草酸“被其他药物影响”。该发现对于指导甘草酸静注制剂的临床安全用药及有效规避风险十分重要。
(李川、贾伟伟翻译撰稿)
原文出处:
Dong JJ, Olaleye OE, Jiang RR, et al. Glycyrrhizin has a high likelihood to be a victim of drug-drug interactions mediated by hepatic OATP1B1/1B3 [J]. Br J Pharmacol, 2018, 175(17): 3486-3503.
其他参考文献:
[1] Feng XC, Ding LQ, Qiu F. Potential drug interactions associated with glycyrrhizin and glycyrrhetinic acid [J]. Drug Metab Rev, 2015, 47(2): 229-238.
[2] Zhang AJ, Li QS, He X, et al. Interactions between transporters and herbal medicines/drugs: a focus on hepatoprotective compounds [J]. Curr Drug Metab, 2015, 16(10): 911-918.
[3] Wu X, Ma J, Ye Y, et al. Transporter modulation by Chinese herbal medicines and its mediated pharmacokinetic herbal-drug interactions [J]. J Chromatogr B, 2016, 1026: 236-253.



